Sindrome de Bart.
El sÃndrome de Bart es un desorden genético caracterizado por ausencia localizada de la piel (presente al nacimiento), epidermólisis ampollosa (EA) y alteraciones ungueales. Es una herencia autosómica dominante, causada por mutación en el gene del colágeno tipo VII en el cromosoma 3p.
EtiologÃa del SÃndrome de Bart.
Christ et al. determinaron las bases genéticas de esta enfermedad al comprobar que se presentaba una mutación en el brazo corto del cromosoma 3p, por una transición G-A en el exón 73, que lleva a una sustitución de glicina- arginina, en el dominio del colágeno tipo VII (COL7A1). La etiologÃa de la ausencia congénita localizada de piel permanece en discusión, aunque para algunos autores puede obedecer a trauma intrauterino, o a adherencias de bandas de lÃquido amniótico y que podrÃan corresponder a manifestaciones in útero de la epidermolisis ampollosa. Duran–McKinster et al plantearon que se podrÃa deber a un mosaicismo segmentario, con base en las manifestaciones clÃnicas observadas en los pocos pacientes reportados en la literatura, pero especialmente por la distribución de las áreas con ausencia de piel que predominan en las extremidades inferiores y que siguen un patrón en forma lineal correspondiente con las lÃneas de Blaschko.
En el año 2004, analizando las caracterÃsticas de las lesiones cutáneas de cinco hermanos con SÃndrome de Bart, que en algunos sitios de las extremidades cursan como grandes zonas de aplasia y en otras como las ampollas tÃpicas de la epidermolisis ampollosa, los autores sugirieron que esto podrÃa obedecer a que como en las enfermedades cutáneas autosómicas se pueden presentar mutaciones post-cigóticas, que se traducen en el paso de las células somáticas a una condición homocigótica, los diferentes grados de afección cutánea observadas en el sÃndrome de Bart se podrÃan explicar por pérdida de la heterocigocidad, en los casos con manifestaciones cutáneas extensas, mientras que las leves corresponderÃan a un fenotipo no mosaico.
CaracterÃsticas clÃnicas.
Las principales manifestaciones clÃnicas incluyen:
1- Ausencia congénita localizada de piel de predominio en miembros inferiores, frecuentemente bilateral, de distribución simétrica, bordes mal definidos y que siguen un patrón en forma de S desde las rodillas hasta la superficie anterolateral de las piernas, cuellos de pié y pies, incluyendo los dedos y en algunos casos las plantas.
2- Malformaciones ungueales como ausencia congénita ungueal, distrofia ungueal o pérdida espontánea de las uñas.
3- Lesiones ampollosas de la piel y las mucosas.
Una variante de la epidermolisis ampollosa de rara presentación llamada epidermólisis ampollosa juncional tipo Herlitz, que también se relaciona con mutaciones, pero en esta caso para en el gen que codifica la laminina 5, se ha reportado en casos de SÃndrome de Bart, siendo pésimo su pronóstico. También hay reportes de SÃndrome de Bart asociado a otras anomalÃas congénitas, dentro de ellas la atresia pilórica.
Tratamiento.
El tratamiento del SÃndrome de Bart es principalmente sintomático, incluye la administración de antibióticos tópicos y la oclusión de las lesiones con gasas vaselinazas, pudiéndose afirmar en términos generales, que el pronóstico de los pacientes es bueno y que las lesiones tienden a mejorar con la edad. Cuando se presenta con atresia pilórica, el manejo quirúrgico de esta malformación se debe hacer con especial cuidado y dependiendo de si hay o no, una buena respuesta de las lesiones cutáneas al tratamiento.
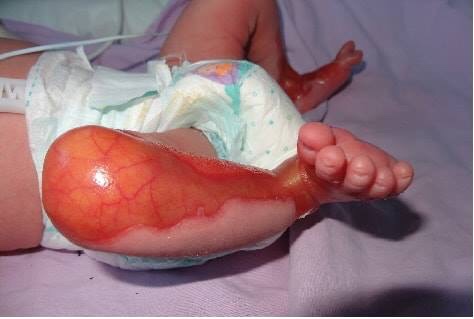
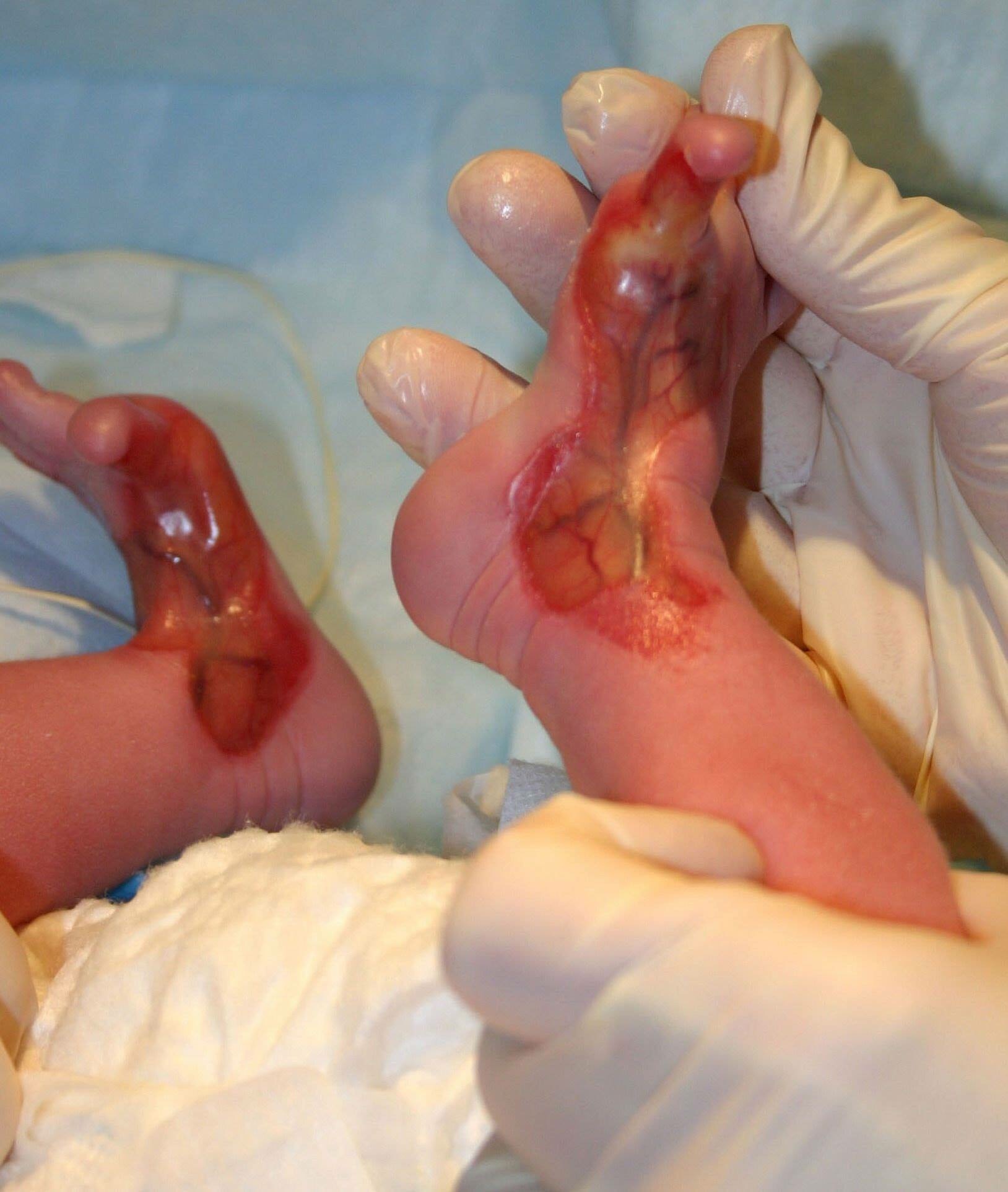
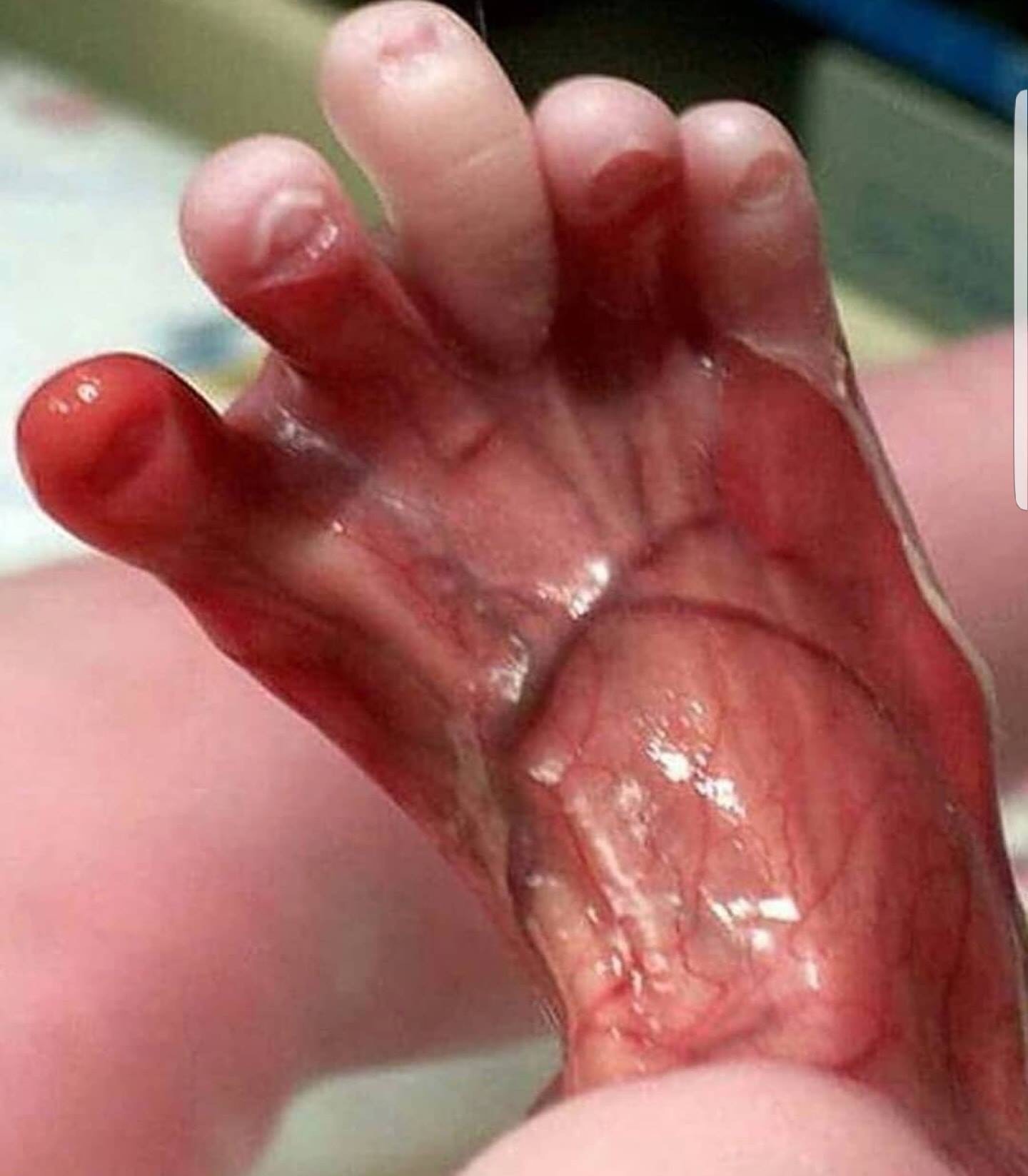
Sindrome de Bart.
bibliografÃa
1. Bartter FC, Pronove P, Gill JR. Hyperplasia of the juxtaglomerular complex with hyperaldosteronism and hypokalemic alkalosis. American Journal of Medicine. 1962. 33:811-828.
2. Seyberth HW. An improved terminology and classification of Bartter-like syndromes. Nat Clin Pract Nephrol. 2008 Oct. 4(10):560-7. [Medline].
3. Bokhari SRA, Mansur A. Bartter Syndrome. 2017 Jun. [Medline]. [Full Text].


